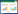 图 1 催化剂的XRD表征图
Fig.1 XRD characterization of catalyst
图 1 催化剂的XRD表征图
Fig.1 XRD characterization of catalyst
CHANG Xiaoning,YANG Ruiqin,CHEN Shuyao,et al.Effect of grain size of ZnZrO2 on preparation of low carbon olefin by CO2 hydrogenation[J].Journal of Zhejiang University of Science and Technology,2024,36(01):68-76.[doi:10.3969/j.issn.1671-8798.2024.01.008]

如何控制CO2过量排放及如何实现能源清洁性都对经济的可持续发展至关重要[1]。在2020年中央经济工作会议上,中国提出了2030年实现“碳达峰”的计划,目标是使CO2排放不再增加,并逐渐回落; 同时为了实现CO2排放量和吸收量相抵消,实现CO2零排放,计划在2060年实现“碳中和”,该计划旨在确保工业发展的同时,有效应对全球气候变化的挑战[2]。CO2是C家族中最大的碳源,具有结构简单、无毒等优点,具有良好的应用前景和经济效益。目前对CO2的研究主要集中在CO2加氢制备高附加值化学品方面,特别是低碳烯烃,这已成为C1化学领域的热点课题[3-4]。乙烯、丙烯、丁烯合称为低碳烯烃(C=2-C=4),在化工行业中有着广泛的应用,可用于制备高分子或其他化合物。工业上,低碳烯烃的生产主要采用重油催化裂解技术[5],这不但会造成能源浪费并且会造成环境恶化,而CO2氢化是合成低碳烯烃的非石油路径,通过该路径可以有效地将CO2转化为小分子烯烃并且合成路线是环保的,是符合新时代中国发展的可行路线。因此,深入研究CO2加氢生产低碳烯烃的技术,对实现CO2的有效利用,减轻石化企业对石化工业的依赖具有非常重要的现实意义[6]。
CO2加氢制备低碳烯烃法的核心是寻找一种结构稳定、催化性能优良的催化剂。对此,Bao等[7-8]提出了氧化物/沸石体系的催化剂设计理念。该体系将CO活化和C=C耦合分离到两种不同类型的活性位点上,这是一种在CO加氢制低碳烯烃中超过费托合成碳链增长机理极限(Anderson-Schulz-Flory,ASF)的有效策略,对所有烃的选择性可高达94%,这一理念同样也适用于CO2的氢化反应。目前通常采用金属催化剂和分子筛复合的方式进行CO2的氢化反应。例如,Li等[9]成功制备出ZnZrO2催化剂,与SAPO -34分子筛组成双功能催化剂后在320 ℃、5.0 MPa等条件下使得CO2可以氢化成低碳烯烃,对碳氢化合物的选择性达到80%以上。
在使用双功能催化剂进行CO2加氢反应的研究中,金属氧化物的结构效应是非常重要的[10],因为金属氧化物组分承担CO2的活化部分。由于存在逆水煤气变换反应,CO2加氢反应的过程常伴随着大量的副产物CO[11],金属氧化物的特性调控对CO2和CO加氢制备高附加值化学品已经受到了广泛关注。Lu等[12]将不同晶粒尺寸的In2O3和SAPO -34相结合用于CO2加氢反应,发现二氧化碳转化率和低碳烯烃选择性随In2O3晶粒尺寸的改变发生变化。Liu等[13]研究了纳米二氧化锆与HZSM-5沸石用于合成气生成芳香族反应,发现二氧化锆表面氧空位数目随着晶粒尺寸的缩小而增多,氧空位数量增多对金属催化剂活化能产生更高的迁移率,从而使金属催化剂更好地参与烃类氧化,并对催化活性有显著提升,这有利于CO通过二氧化锆向甲醇转变。Li等[14]将ZnO/SAPO -34双功能催化剂用于合成气转换反应中,发现合成气转化率和低碳烯烃选择性随着氧化锌晶粒尺寸的减小而逐渐增强。尽管目前国内外对双功能催化剂在CO2加氢反应的研究较多,但双功能催化剂中ZnZrO2晶粒尺寸对CO2氢化的活性和产物分布方面的研究尚不多见。因此,本研究通过改变焙烧温度使金属催化剂ZnZrO2的晶粒尺寸发生改变; 并在ZnZrO2/SAPO -34双功能催化剂的作用下,研究ZnZrO2的晶粒尺寸对CO2加氢制备低碳烯烃反应的影响。
1 试验部分1.1 试验材料硝酸锌(Zn(NO3)2·6H2O)、硝酸锆(Zr(NO3)4·5H2O)、碳酸铵((NH4)2CO3)、乙醇、磷酸(H3PO4,质量分数为85%)、勃目石(Al2O3,质量分数为70%)、三乙胺,均购自上海凌峰化学试剂有限公司; 四乙基氢氧化铵购自上海阿达玛斯(Adamas Beta)有限公司; 气相二氧化硅购自国药化学试剂有限公司; 反应气体购自杭州民星化工科技有限公司; 所用化学试剂均为分析纯。
1.2 催化剂制备1.2.1 锌锆催化剂锌锆催化剂采用共沉淀法制得。设定锌锆摩尔比为13:87,将一定量的硝酸锌、硝酸锆溶于100 mL去离子水中,常温搅拌至完全溶解,溶液标记为A溶液; 将一定量(NH4)2CO3溶于另一100 mL去离子水中,搅拌至完全溶解,溶液标记为B溶液。采用双管共同滴定的方法将A、B两溶液缓慢滴入100 mL恒温80 ℃的去离子水中,溶液的pH值控制在8.0左右,持续搅拌4 h后将烧杯拿出,冷却至室温后用去离子水洗涤至中性,然后在100 ℃烘箱中干燥12 h,将干燥后的催化剂以2.5 ℃/min的升温条件在不同的温度下焙烧3 h,冷却至室温后便制备出一系列不同粒径的锌锆固溶体样品,将样品命名为ZnZrO2-x(其中x代表焙烧温度)。
1.2.2 SAPO-34分子筛催化剂采用水热合成法制备SAPO-34分子筛。设定样品Al2O3、P2O5、SiO2、三乙胺、四乙基氢氧化铵、H2O的摩尔比为:1.0:1.0:0.2:3.0:2.5:60。首先,在室温条件下,将勃目石倒入去离子水中,持续搅拌30 min,在持续搅拌时分别滴加一定量的磷酸和气相二氧化硅,搅拌2 h后再分别滴加模板剂三乙胺和四乙基氢氧化铵直至形成凝胶(约20 min)。之后,将凝胶密封在100 mL特氟龙内衬不锈钢容器中,在200 ℃自生压力下结晶24 h。经过离心、洗涤后在100 ℃烘箱中干燥12 h,干燥后以2.5 ℃/min的升温条件在550 ℃条件下煅烧6 h以去除模板剂。
1.2.3 ZnZrO2-x/SAPO-34双功能催化剂将制备好的ZnZrO2-x催化剂和SAPO-34分子筛以质量比为1:1进行物理研磨并进行压片处理,最终造粒成20~40目大小。将样品命名为ZnZrO2-x/SAPO-34。
1.3 催化剂性能评价在固定床反应器上进行CO2加氢制备低碳烯烃的反应性能评价。反应管的内径尺寸为6.8 mm,称量1.0 g 20~40目ZnZrO2-x/SAPO-34催化剂,放置在反应管的中央恒温区域,在催化剂的上下两端用石英棉固定,催化剂在常压下使用体积分数为99.9%的H2(流速40 mL/min)还原催化剂。反应器冷却后,通入Ar、CO2、H2的混合气体,体积比为1:6:18,吹扫20 min,吹扫结束后,设定一定的压力和温度进行升压和升温操作。当反应温度与压力稳定后,设置反应气体的流速为40 mL/min,反应时间为6 h。反应后管线和阀门被加热并保持在120 ℃,以避免产物冷凝。所有数据均在反应1 h稳定之后获得。在反应过程中每隔60 min取1次样,进行样品产物分析。所有气体产物使用安捷伦色谱柱(Agilent HP-Plot/Q,配备火焰离子化检测仪)和碳分子筛填充色谱柱(TDX-01,配备热导检测器)进行在线分析。CO2转化率和目标产物的选择性计算公式如下:
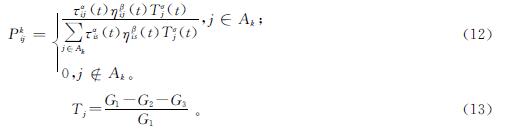
式(1)、式(2)和式(3)中:XCO2为CO2的转化率; Fin为进料气体的入口气流的峰面积; CCO2,in为进气的CO2摩尔浓度; Fout为气体出口气流的峰面积; CCO2,out为出口气体的CO2摩尔浓度; Si为i组分选择性; ni为产物中i组分中C原子的数量; Ci,out为出口气体i组分摩尔浓度; yi为i组分的实际输出。
2 结果和讨论2.1 催化剂的物相分析催化剂的XRD(X-ray diffraction,X射线衍射)表征图见图1,SAPO-34样品的主要衍射峰2θ分别为9.5°、12.8°、20.6°、25.9°、30.7°,制备的产物结晶度高且无杂峰,表明水热合成法成功合成了沸石[15]。通过对比标准卡片,ZnZrO2-x样品在30.3°、35.2°、50.6°、60.2°、63.0°、74.5°处分别对应为四方相ZrO2(t-ZrO2)的101、110、200、211、202、220平面衍射峰,ZnZrO2-x结晶度随着焙烧温度升高而增加,说明金属颗粒会随着焙烧温度的升高而聚集,与SEM(scanning electron microscope,扫描电子显微镜)表征结果一致。在t-ZrO2中碱性位占主导地位,此外ZrO2是p型半导体,容易产生氧空穴,作为催化剂载体可与活性组分产生较强的相互作用[16]。ZnZrO2-x催化剂中均没有观察到ZnO的特征衍射峰,表明ZnO并非独立的,均匀分散在ZnZrO2催化剂中,形成了ZnZrO2固溶结构[17]。
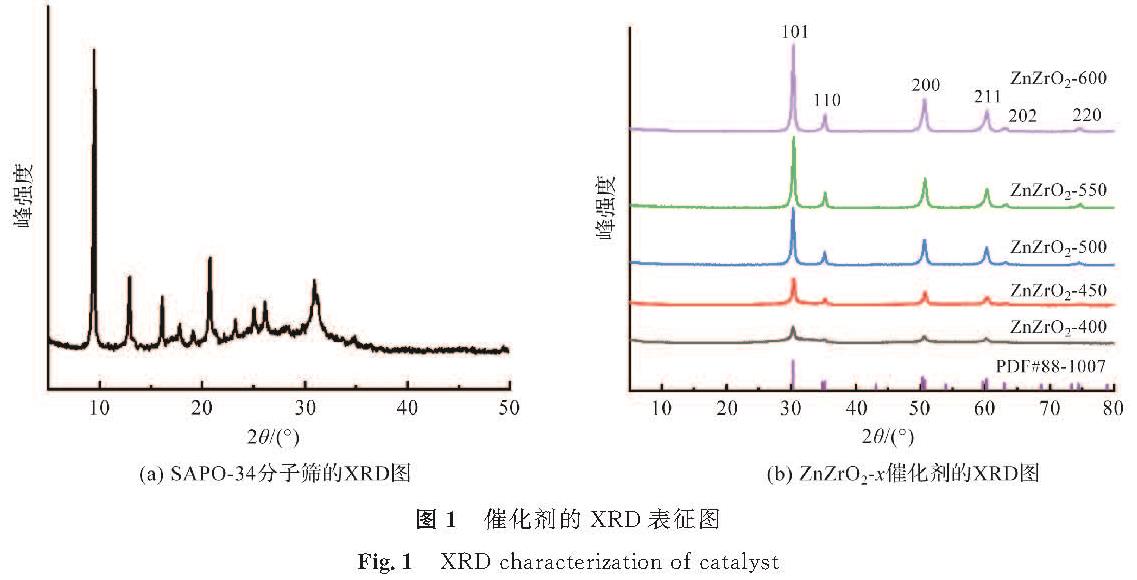
图1 催化剂的XRD表征图
Fig.1 XRD characterization of catalyst
利用谢勒(Scherrer)公式,在ZnZrO2(101)XRD峰的半峰全宽最大值处确定了ZnZrO2-x样品的平均晶粒尺寸
D=(Kr)/(βcosθ)。(4)
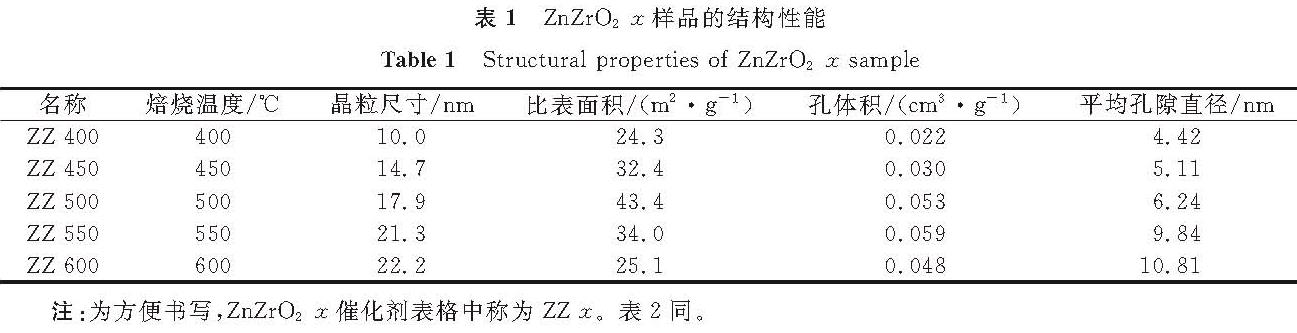
式(4)中:K为谢勒常数(球形粒子为0.89,立方离子为0.943); r为X射线波长,一般为0.154 056 nm; β为实测样品衍射峰半高宽度或者积分宽度。式(4)适用于晶粒尺寸大小在1~100 nm范围内的金属催化剂。随着焙烧温度的不断增加,ZnZrO2样品的粒径从10.0 nm逐渐增加到22.2 nm。ZnZrO2-x样品的结构性能见表1。
表1 ZnZrO2-x样品的结构性能
Table 1 Structural properties of ZnZrO2-x sample
2.2 催化剂的比表面积及孔结构分析
N2-吸附脱附曲线有助于判断样品的孔径和孔隙率。不同焙烧温度对ZnZrO2催化剂的N2-吸附脱附曲线有较大的影响[18],ZnZrO2-x催化剂的N2-吸附脱附曲线如图2所示。所有样品均表现出Ⅳ型等温线,在ZnZrO2-x样品的结构中存在介孔,介孔结构有利于反应物和产物的传质扩散过程。如表1所示,焙烧温度从400 ℃不断增加到500 ℃,ZnZrO2晶粒尺寸从10.0 nm增加到17.9 nm,空隙率增加导致催化剂的比表面积从24.3 m2/g增加到43.4 m2/g; 随着焙烧温度从500 ℃增加到600 ℃,ZnZrO2晶粒尺寸从17.9 nm增加到22.2 nm,比表面积却从43.4 m2/g减少到25.1 m2/g,这可能是由于焙烧温度过高导致金属颗粒团聚进而引起比表面积的下降。ZnZrO2晶粒尺寸和比表面关系如图2(c)所示。
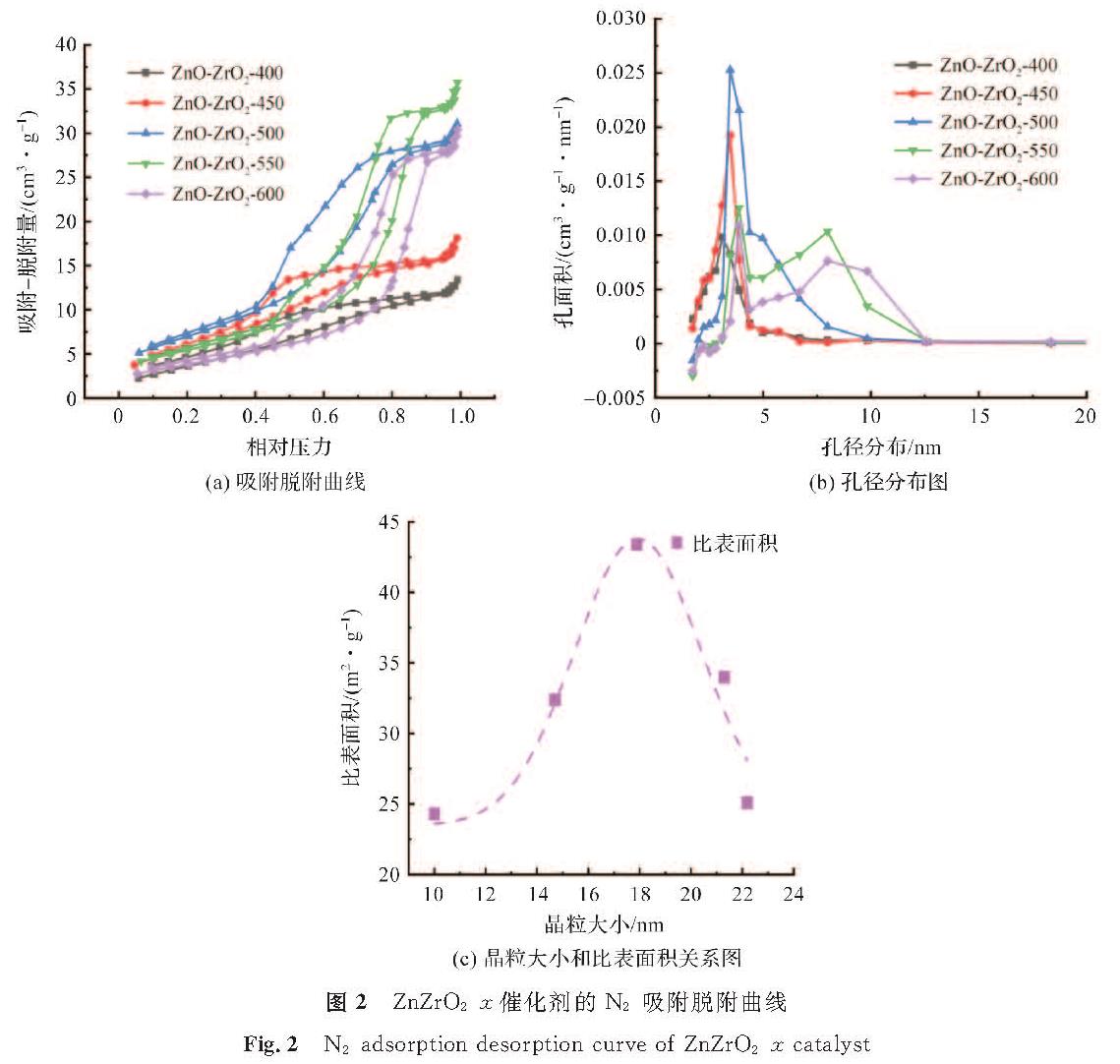
图2 ZnZrO2-x催化剂的N2-吸附脱附曲线
Fig.2 N2-adsorption desorption curve of ZnZrO2-x catalyst
随着焙烧温度从400 ℃增大到500 ℃,催化剂孔体积逐渐增大,孔径的峰高在不断增大,说明介孔的数量在不断增加,当焙烧温度从500 ℃增大到600 ℃时,孔径的峰高降低,这说明过高的焙烧温度可能会导致孔隙的坍塌现象。焙烧温度的升高对比表面积的影响可以说明,随着ZnZrO2粒径的增大,比表面积呈火山变化趋势。ZnZrO2-500催化剂与其他不同焙烧温度催化剂相比具有最大的比表面积,该金属催化剂与分子筛组成了双功能催化剂,这可能是在催化反应中CO2转化率最高的原因之一。
2.3 催化剂的红外分析催化剂的FT-IR(Fourier transform infrared spectoscopy,傅里叶变换红外光谱法)图见图3。通过对比可以发现,金属催化剂ZnZrO2-x在470 cm-1和640 cm-1处金属O—Zn—O的特征振动带强度随着焙烧温度的升高而加强,这是由金属催化剂ZnZrO2颗粒随着焙烧温度的升高不断聚集造成的,其中焙烧温度为600 ℃时,O—Zn—O官能团的吸光率在所有焙烧温度中是最高的,这与XRD、SEM表征结果一致。2 340 cm-1和3 420 cm-1处的特征振动带分别为空气中游离的CO2分子中C=O键的伸缩振动峰和样品保存时吸附水形成的羟基,振动强度随着焙烧温度的增加反而降低,我们猜测在制备过程中,焙烧温度越高的催化剂越不易被外界环境影响。
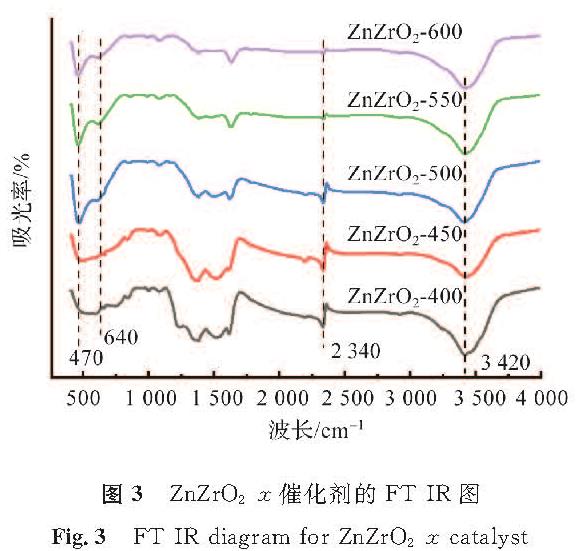
图3 ZnZrO2-x催化剂的FT-IR图
Fig.3 FT-IR diagram for ZnZrO2-x catalystZnZrO2-x
2.4 催化剂的形貌和表面元素分析
扫描电镜可以观察到催化剂的形貌和表面元素分布,催化剂的SEM图和EDS(energy dispersive spectrometer,能量色散光谱仪)图见图4。
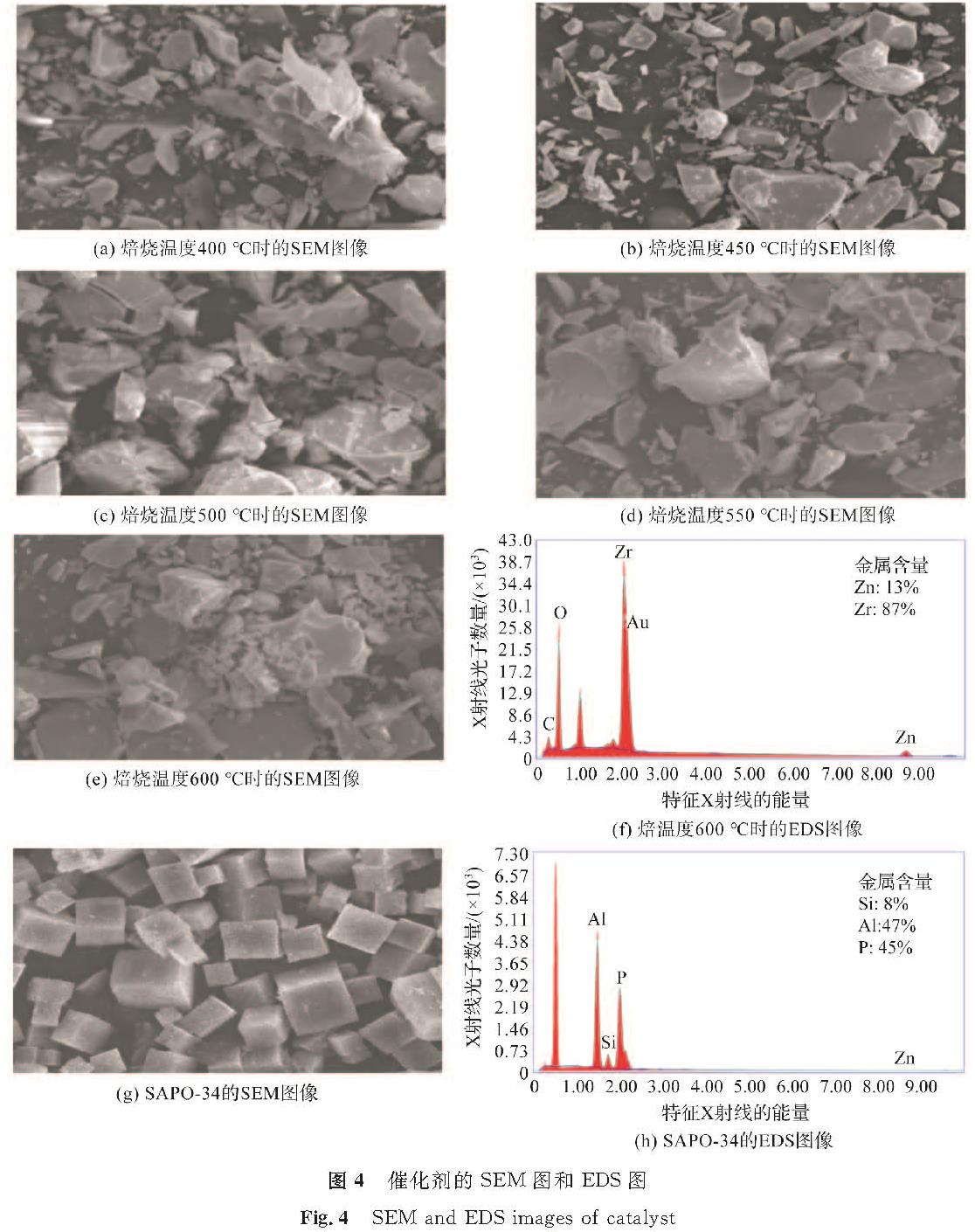
图4 催化剂的SEM图和EDS图
Fig.4 SEM and EDS images of catalyst
图4(g)合成的SAPO-34沸石具有CHA(Canadian Hydrogen Association,加拿大氢协会)拓扑结构[19],平均颗粒大小在2~5 μm之间; ZnZrO2催化剂由许多不均匀的颗粒组成,进一步通过EDS表征分析了ZnZrO2催化剂和SAPO-34的元素摩尔比,结果表明元素比例符合固溶体制备时设计的比例,证明催化剂制备成功。当焙烧温度过低时,ZnZrO2催化剂的颗粒较小且聚集不明显,随着焙烧温度的不断增加,ZnZrO2催化剂的颗粒逐渐变大,并且金属颗粒聚集明显,这也是XRD表征中结晶度不断增大的原因。
2.5 催化剂的酸性分析在二氧化碳转化过程中,表面基本性质非常重要,这可以决定二氧化碳的活化途径,并进一步影响其催化性能。为了分析不同焙烧温度对金属催化剂酸性的影响,对金属催化剂进行了NH3-TPD(ammonia temperature-programmed desorption,氨-程序升温脱附)测试,ZnZrO2-x催化剂的NH3-TPD图见图5。催化剂以170~200 ℃为中心存在一个氨气解吸峰,对应于催化剂的弱酸位点,在测试温度为450~600 ℃时,催化剂与测试气体发生了反应。由于设定CO2加氢的反应温度为380 ℃,故对催化剂在400 ℃之前弱酸位点进行分析,ZnZrO2-400、ZnZrO2-500、ZnZrO2-550催化剂中的酸强度从大到小排序为:ZnZrO2-500、ZnZrO2-550、ZnZrO2-400。随着ZnZrO2晶粒尺寸从10 nm增加到17.9 nm,氨气的吸附量显著增加; 晶粒尺寸从17.9 nm继续增加到21.3 nm,氨气的吸附量反而下降。这些结果进一步表明,较大粒径的金属氧化物颗粒具有更多的弱酸量,但颗粒增大到一定值时,会破坏催化剂的酸性,从而导致弱酸量下降。
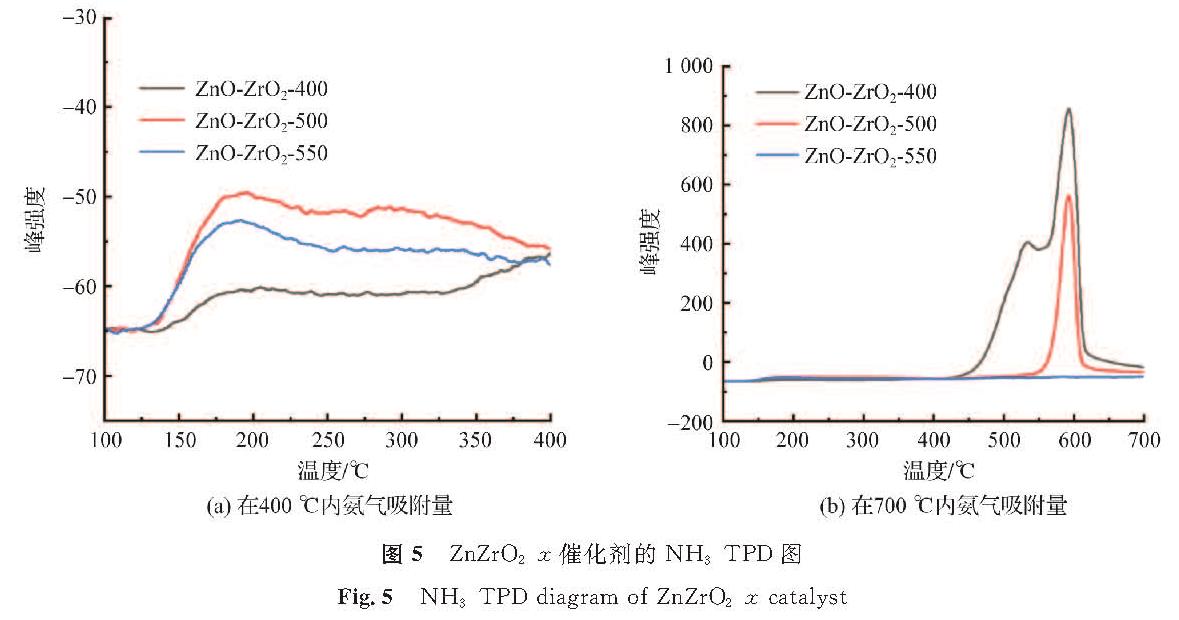
图5 ZnZrO2-x催化剂的NH3-TPD图
Fig.5 NH3-TPD diagram of ZnZrO2-x catalyst
2.6 催化剂的化学吸附分析
氧化物/沸石催化剂为CO2加氢生成碳氢化合物提供了两种活性位点。CO2首先在金属氧化物处形成C1加氧物(甲醇或CHxO物种),然后C1加氧物移动到沸石的酸性位置,并通过烃池机制转化为碳氢化合物,而对于CO2在ZnZrO2催化剂上加氢,人们普遍认为二氧化碳的吸附性对H2的活化能力至关重要。因此,为了解ZnZrO2-x样品的二氧化碳吸附能力,对催化剂进行了CO2-TPD(CO2 temperature-programmed desorption,二氧化碳-程序升温脱附)表征测试,试验结果如图6所示。所有催化剂在位于120~250 ℃处的α峰对应于物理吸附的二氧化碳,大于450 ℃时催化剂与测试气体发生了反应,与NH3-TPD的测试结果一致。焙烧温度从400 ℃提高到500 ℃,在α峰位置处ZnZrO2-500催化剂在所有样品中显示出较强的峰,这可能是较大的比表面积导致CO2吸附量的增加。
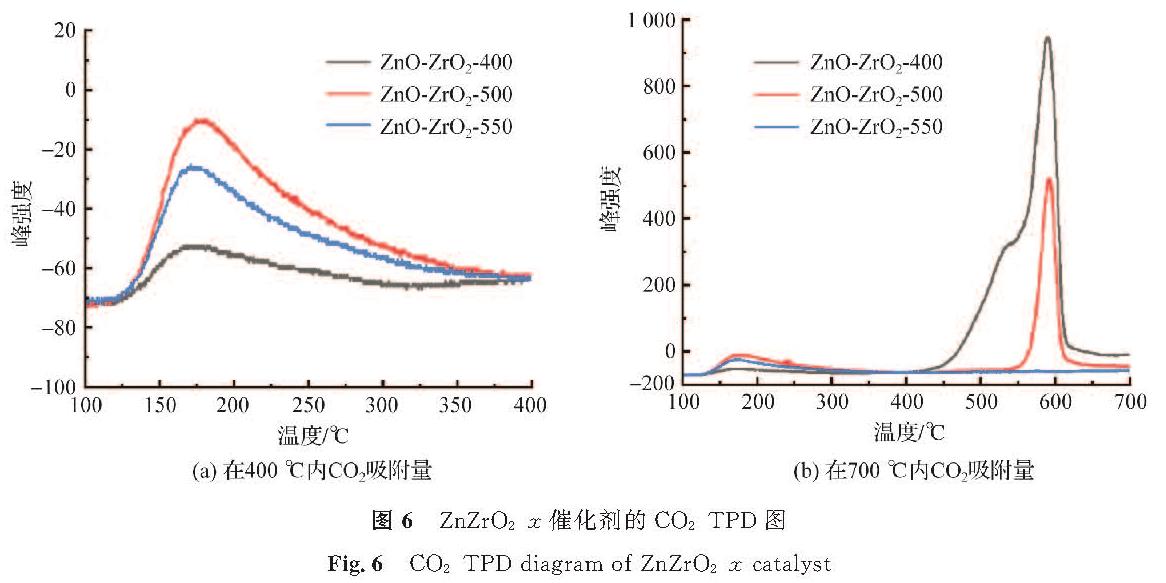
图6 ZnZrO2-x催化剂的CO2-TPD图
Fig.6 CO2-TPD diagram of ZnZrO2-x catalyst
2.7 催化活性评价
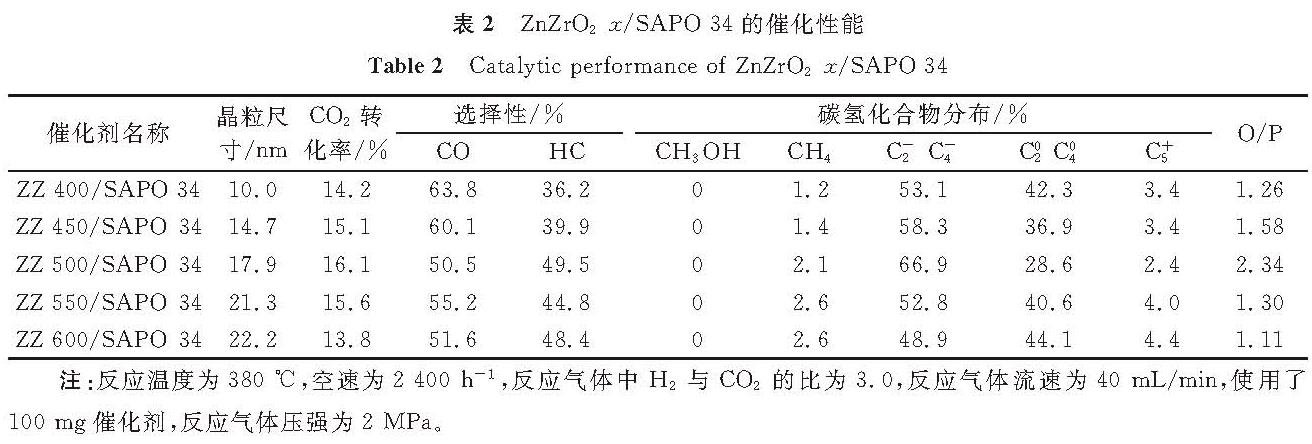
将ZnZrO2-x催化剂与SAPO-34沸石以相同质量比物理混合,作为CO2加氢反应的双功能催化剂。ZnZrO2-x/SAPO-34的催化性能见表2。晶粒尺寸的改变对C+5烃类和副产物甲烷的选择性影响较小; 然而,它对低碳烯烃(C=2-C=4)和石蜡(C02-C04)的选择性有很大的影响。例如,当ZnZrO2晶粒尺寸从10.0 nm逐渐增大到17.9 nm时,C=2-C=4的选择性增强,低碳烯烃与石蜡的比值(O/P)逐渐增大,并达到最大值66.9%与2.34; 但随着氧化物颗粒粒径的进一步增加,两者均逐渐降低。随着ZnZrO2晶粒尺寸的增加,石蜡的选择性呈现出先减弱后增强的变化规律。C=2-C=4的选择性和O/P比值的变化可以归因于ZnZrO2的酸性性质,因为烯烃的催化氢化可以发生在酸性位点,而17.9 nm的ZnZrO2纳米颗粒具有较多的酸量,适宜的酸量大小会防止烯烃的进一步催化氢化,利于低碳烯烃的生成; 当ZnZrO2晶粒小于或超过17.9 nm时,反应产物烯烃选择性减弱,O/P比值减小,证明通过改变金属催化剂的焙烧温度改变了催化剂晶粒尺寸,并影响了金属颗粒表面的物化性质,实现了对目标产物的有效调控。
表2 ZnZrO2-x/SAPO-34的催化性能
Table 2 Catalytic performance of ZnZrO2-x/SAPO-34
ZnZrO2晶粒尺寸与转化率关系如图7所示,ZnZrO2晶粒尺寸会影响CO2的转化率和CO副产物的选择性。在ZnZrO2晶粒尺寸为17.9 nm时,CO2转化率达到最高的16.1%; 但当晶粒尺寸从17.9 nm增加到22.2 nm时,CO2转化率则显著下降。通过CO2-TPD分析可知,这是由于ZnZrO2-500催化剂具有更好的CO2吸附性能,这对H2的活化性能至关重要,因此该催化剂表现出优良的催化性能。CO的选择性随着晶粒尺寸的变大而降低,我们猜测,晶粒越大的金属催化剂可能越有利于抑制逆水煤气反应,从而使CO选择性降低。
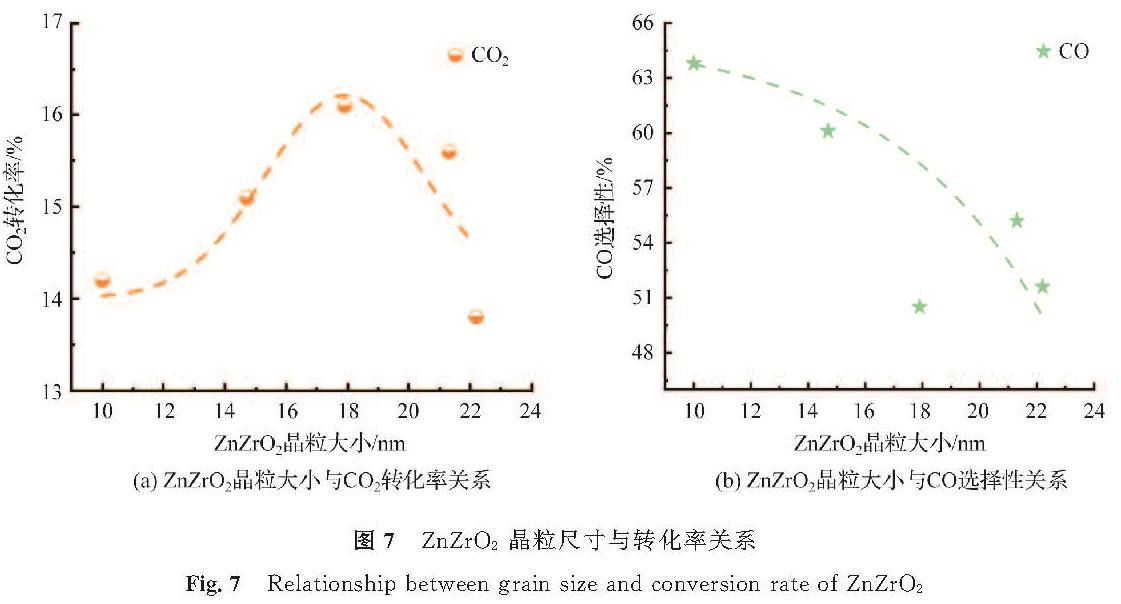
图7 ZnZrO2晶粒尺寸与转化率关系
Fig.7 Relationship between grain size and conversion rate of ZnZrO2
3 结 语
通过改变焙烧温度合成了晶粒尺寸在10.0~22.2 nm的ZnZrO2催化剂,并将不同ZnZrO2晶粒尺寸的催化剂与SAPO-34组成双功能催化剂,研究其对CO2加氢反应制备低碳烯烃的影响。目标产物中低碳烯烃的选择性随着ZnZrO2晶粒尺寸的变大而增强,但当晶粒大于17.9 nm时,选择性则随着晶粒尺寸的进一步增加而减弱。适量大小的ZnZrO2晶粒具有更大的比表面积和更多的酸量,以及更好的CO2吸附性能,为CO2和H2的活化提供了更多的活性位点。ZnZrO2最佳晶粒尺寸为17.9 nm,该尺寸下CO2选择性达到16.1%,低碳烯烃选择性为66.9%。综上所述,适宜的ZnZrO2晶粒尺寸可以促进二氧化碳加氢反应,并降低烯烃进一步发生反应。这些结果为设计出尺寸更合适、催化性能更高效的金属催化剂并应用于CO2加氢反应提供了试验数据。
- [1] 周桂林,艾鑫.CO2加氢逆水煤气变换(RWGS)催化剂研究进展[J].重庆工商大学学报(自然科学版),2023,40(1):9.
- [2] 王林霞.积极稳妥推进“碳达峰碳中和”解码中国绿色低碳发展之路[J].陕西行政学院学报,2023,37(1):13.
- [3] 成康,张庆红,康金灿,等.二氧化碳直接制备高值化学品中的接力催化方法[J].中国科学:化学,2020,50(7):745.
- [4] 中国石化有机原料科技情报中心站.华侨大学的CO2直接制低碳烯烃催化剂研究获进展[J].石油炼制与化工,2022,53(10):20.
- [5] 侯凯军,高金森,马安,等.重油催化裂解制低碳烯烃工艺技术研究进展[J].应用化工,2022,51(11):3279.
- [6] 李泽龙,曲圆直,王集杰,等.CO2加氢高选择性制备低碳烯烃、芳烃[C]//第一届全国二氧化碳资源化利用学术会议.天津:中国化学会,2019:1.
- [7] LI J, YU T, MIAO D Y, et al. Carbon dioxide hydrogenation to light olefins over ZnO-Y2O3 and SAPO-34 bifunctional catalysts[J].Catalysis Communications,2019,129:105711.
- [8] JIAO F, LI J J, PAN X, et al. Selective conversion of syngas to light olefins[J].Science,2016,351(6277):1066.
- [9] ZHANG X, ZHANG A, JIANG X, et al. Utilization of CO2 for aromatics production over ZnO/ZrO2-ZSM-5 tandem catalyst[J].Journal of CO2 Utilization,2019,29:142.
- [10] 范兴其.双金属氧化物/SAPO-34串联催化剂的制备及CO2加氢反应研究[D].贵阳:贵州大学,2021.
- [11] 乔婷婷,栗怡,赵玺乐等.逆水煤气变换反应催化剂的研究进展[J].农村经济与科技,2020,31(18):274.
- [12] LU S Y, YAN H Y, ZHOU Z X, et al. Effect of In2O3 particle size on CO2 hydrogenation to lower olefins over bifunctional catalysts[J].Chinese Journal of Catalysis,2021,42(11):2039.
- [13] LIU J G, HE Y R, YAN L L, et al. Nano-sized ZrO2 derived from metal-organic frameworks and their catalytic performance for aromatic synthesis from syngas[J].Catalysis Science and Technology,2019,9:2983.
- [14] LI N, JIAO F, PAN X L, et al. Size effects of ZnO nanoparticles in bifunctional catalysts for selective syngas conversion[J].ACS Catalysis,2019,9(2):962.
- [15] LU P, RISWAN M, CHANG X N, et al. Hydrogenation of CO2 to light olefins on ZZ/MnSAPO-34@Si-2:effect of silicalite-2 seeds on the acidity and catalytic activity[J].Fuel,2022,330:125470.
- [16] 张秀娟,郑婷婷,王成雄,等.ZrO2基催化剂在天然气汽车尾气处理中的应用研究进展[J].稀有金属,2022,46(12):1633.
- [17] Tong M L, GAPU C L, CHANG X N, et al. Tandem catalysis over tailored ZnO-ZrO2/MnSAPO-34 composite catalyst for enhanced light olefins selectivity in CO2 hydrogenation[J].Microporous and Mesoporous Materials,2021,320:111105.
- [18] 刘昊霖,叶超,许友生,等.生物炭-Fe2O3的制备及其脱除焦油模型化合物研究[J].浙江科技学院学报,2022,34(6):543.
- [19] 尚蕴山,刘意,陈昊源,等.水热处理对SAPO-34分子筛在合成气一步法制烯烃反应中性能的影响[J].天然气化工:C1化学与化工,2022,47(3):75.